단백질 구조는 아미노산 서열 시퀀스로 표현되는 1차 구조로 시작해, 알파헬릭스, 베타시트로 구성되는 로컬한 구조 정보를 표현하는 2차 구조, 글로벌한 좌표 정보를 표현하는 3차 접힘(folding) 구조, 그리고 다수의 단백질 체인들이 결합되는 4차 구조로 이루어져 있다. 서로 다른 단백질들이 산소를 운반하는 헤모글로빈 단백질이 될지, 눈 근육에 있는 미오신 단백질이 될지는 아미노산 서열 시퀀스와 이로 인한 단백질 접힘으로 결정된다. 이번 글에서는 단백질의 모든 것을 결정하는 단백질 구조의 4단계에 대해 알아보겠다.

아미노산
단백질의 구조에 대해 논하기 전에, 단백질을 구성하는 기본 요소인 아미노산(amino acid; residue)에 대해 먼저 알아보겠다. 지구상에 존재하는 모든 종류의 단백질은 단 20개의 아미노산으로 구성된다. 이 아미노산은 앞서 말했듯 단백질의 가장 기본이 되는 구성 요소이고, 쉽게 말해 자연어의 단어가 하는 것과 같은 역할을 한다. 아래 그림을 보면, 20개 종류의 아미노산을 한 번에 파악할 수 있다.
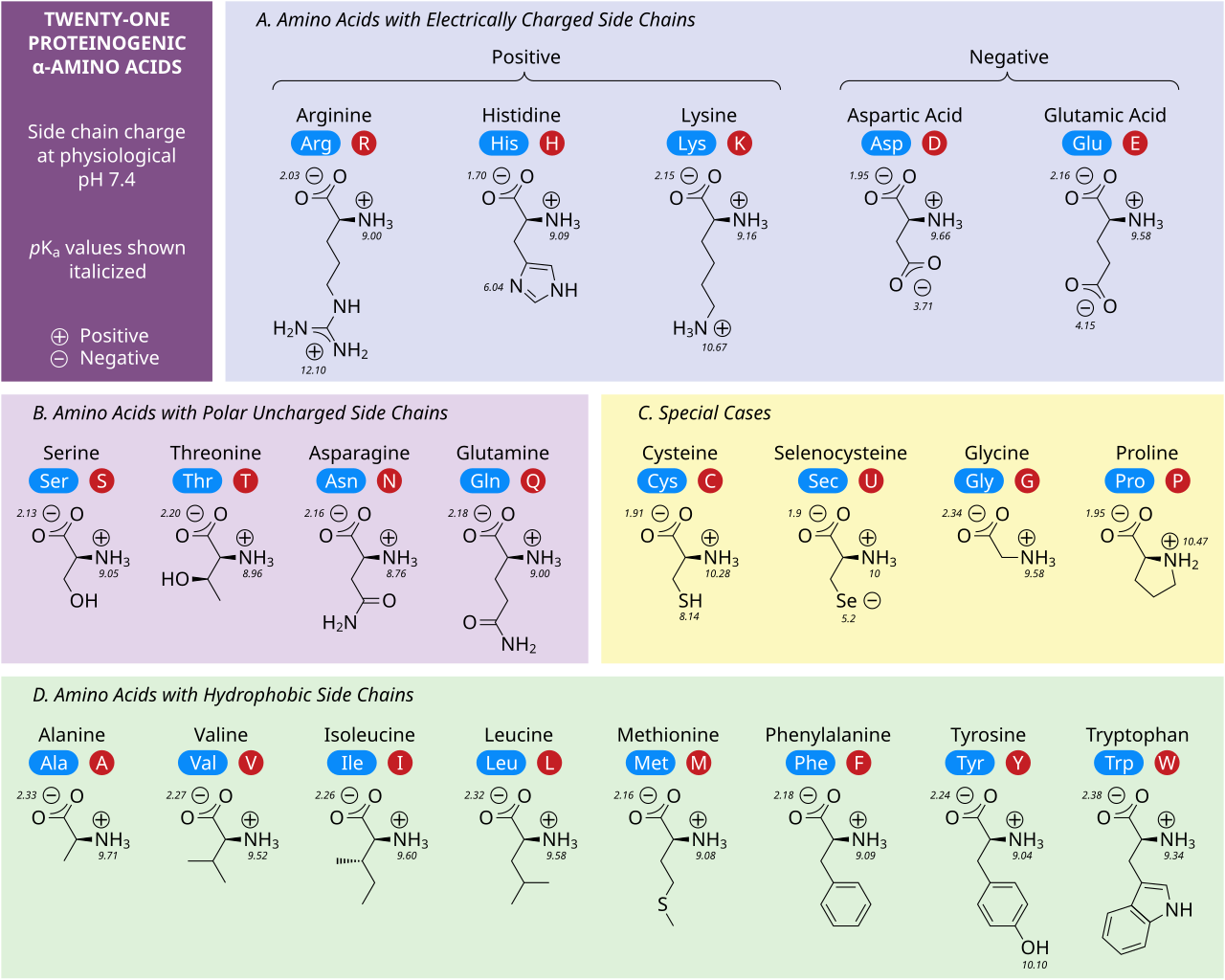
구체적인 분자 구조를 외울 필요는 절대 없고 (나도 모름), 각 아미노산이 이름과 분자 구조에 따른 고유한 특성을 가지고 있고, 줄여서 알파벳 세 글자(파란색) 혹은 한 글자(빨간색)으로 표현된다는 사실만 알고 있으면 된다. 위 아미노산들의 분자 구조를 살펴 보면, 아래와 같은 구조를 모든 분자가 공유하고 있다는 것을 알 수 있다.

각 아미노산은 위와 같은 공통적인 기본적인 구조를 가지고 있다. 구체적으로 설명하면, 중심 탄소가 아미노기(amino group; NH2)와 카복실기(carboxyl group;COOH)와 결합된 공통적인 구조를 가지고 있고, 이것에 각 아미노산의 고유한 특성을 부여하는 R group (side chain)이 붙어 있는 형태이다. 이렇게 아미노산은 R group의 종류에 따라 아래와 같이 고유한 화학적 특성을 가지게 된다.

위 그림을 간단히 살펴보면, 아미노산마다 분자량(molecular weight)도 다 다르고, 극성을 띄는지 띄지 않는지(소수성), 양전하인지 음전하인지의 분자적 특성도 모두 다름을 알 수 있다. 이렇게 서로 다른 아미노산의 특성은 단백질의 접힘(folding)이 결정될 때, 중요한 영향을 미친다. 예를 들어, 극성을 띄지 않는 nonpolar 아미노산들은 물을 싫어하기(소수성) 때문에, 단백질의 내부로 뭉치는 경향이 있는 식이다.
1차 구조 (primary structure)
단백질의 1차 구조는 아래 그림과 같이 아미노산의 시퀀스 서열 그 자체를 의미하고, 자연어로 치면 문장의 역할을 한다. 이 때는 단백질 내부 원자의 위치나 접힘, 로컬한 구조 등을 모두 고려하지 않고 아미노산의 순서 만을 고려한다.

각 아미노산은 본인의 amino group과 옆에 있는 아미노산의 carboxyl group 사이의 peptide bond로 연결된다. 이렇게 연결된 N-C-C- 구조를 단백질의 backbone 구조라고 부른다.

단순해 보일 수 있으나, 여기에는 막대한 정보가 담겨 있다. 각 위치에 20개의 아미노산이 위치할 수 있고 단백질이 수백에서 수천 개의 아미노산 길이를 가질 수 있으므로, 가능한 서열의 수는 천문학적으로 많고, 이러한 막대한 조합의 단백질 종류들은 종마다 개체마다 다른 특성을 가질 수 있게 하는 핵심적 요인이고, 긴 단백질 시퀀스 내에서 하나의 아미노산만 바뀌어도 질병을 유발할 수 있다. 예를 들어, 헤모글로빈 단백질 내의 단 하나의 아미노산 E가 V로 대체되면 겸상 적혈구 질환(Sickle cell disease)을 유발한다. 즉, 단 하나의 아미노산이 단백질의 전체 폴딩 구조를 변환시켜 화학적 성질을 변화하게 만드는 것이다.
2차 구조 (secondary structure)
단백질의 2차 구조는 1차 구조 내의 local segment(예를 들어, 연속된 아미노산 8개 등)가 규칙적이고 반복적인 기하학적 구조로 접히는 것을 의미한다. 이러한 로컬 구조는 단백질의 carboxyl oxygen (C=O)와 amide hydrogen (N-H) 사이의 수소 결합으로 만들어진다.

이러한 2차 구조에는 주로 알파 헬릭스와 베타 시트의 두 가지 형태가 있다. 알파 헬릭스는 위 그림의 오른쪽처럼 수소결합이 나선 축과 평행한 형태로 구성되어 나선형으로 꼬인 구조를 의미한다. 이 수소 결합은 약 3.6개의 아미노산마다 한 개 형성된다. 베타 시트는 그림의 왼쪽처럼 수소 결합이 세그먼트를 가로지르는 형태로 구성되어 두 개 이상의 세그먼트가 나란히 놓인 구조를 의미한다. 이 때, 하나의 세그먼트는 베타 strand라고 표현한다. 마지막으로, 이러한 알파 헬릭스 혹은 베타 시트 모두에 포함되지 않고, 이들을 연결하는 비정형적인 구간을 보통 loop라고 부른다. 이러한 loop는 구조적인 유연성을 부여하여 단백질이 복잡한 3차 구조로 folding할 수 있게 돕는 역할도 하고, 단백질 표면에 주로 노출되어 다른 분자 혹은 단백질과의 상호작용(binding)에 있어 핵심적인 역할을 한다.
3차 구조 (tertiary structure)
단백질의 3차 구조는 이러한 2차 구조들이 모여서 folding하는 완전한 3차원 구조를 의미한다. 이 구조를 통해 단백질은 완전한 기능성을 갖추게 된다. 이를 일어나게 하는 데에는 크게 네 가지 요인이 있다.
- 소수성 효과 (Hydrophobic effect): 위에서 설명한 소수성의 nonpolar side chain이 물을 피하여 단백질 내부로 이동하여 소수성 코어를 형성한다.
- 수소 결합 (Hydrogen bonds): 2차 구조의 골격 외에도 polar side chain 간에 형성된다.
- 이온 결합 (Ionic bonds) / salt bridges: 반대 전하를 띤 side chain, 즉 양전하 side chain과 음전하 side chain 간에 형성된다.
- 이황화 결합 (Disulfide bonds): 두 cysteine 아미노산의 -SH 그룹 간의 공유 결합으로, 3차 구조에서 가장 강력한 결합이다.
이러한 3차 구조는 1차 구조에 의해 전적으로 결정된다. 즉, 단백질을 화학적으로 다시 풀어 놓아도 자발적으로 1차 구조가 내재적으로 가지고 있는 3차 구조로 다시 folding한다는 것이다. 이렇게 1차 구조 시퀀스가 주어졌을 때, 3차 구조를 예측하는 방법론 중 하나가 그 유명한 Alphafold이다.
4차 구조 (quatenary structure)
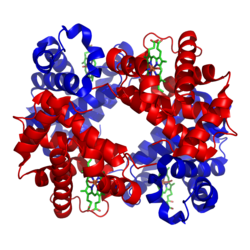
단백질의 4차 구조는 단백질들이 하나의 고립된 체인으로 작동하지 않고, 여러 체인들이 결합하여 더 큰 복합체를 형성한 것을 말한다. 예를 들어, 아래 그림과 같이 헤모글로빈은 4개의 체인으로 구성된 복합체이다. 중요하지는 않지만, 재미를 위해 설명하자면 각 체인 간의 상호작용이 헤모글로빈이 폐에서 산소를 적재하여 조직에서 효율적으로 방출할 수 있도록 도와준다. 첫 번째 체인이 산소와 결합하면 미세한 구조적 변화가 다른 체인으로 전파되어 산소 친화력을 높이고, 네 번째 체인은 첫 번째보다 훨씬 더 높은 친화력으로 산소와 결합하는 식이다.
결론
이번 글의 핵심은 네 가지 구조 모두 궁극적으로 1차 구조에 의해 결정된다는 것이다. 1차 서열에서 4차 구조로 이어지는 이러한 계층적 결정론은, 우리가 단백질의 아미노산 시퀀스를 읽을 수 있다면 원칙적으로 그 형태와 기능에 관한 모든 것을 알 수 있음을 시사한다. 그렇기 때문에 1차 서열로부터 3차 구조를 예측하는 단백질 접힘(Protein folding) 문제는 계산 생물학에서 가장 어려운 과제 중 하나였다. 하지만 Alphafold는 이를 해결했고, 전체 구조 데이터베이스를 공개하기도 하였다. 단백질 구조의 이해는 신약 개발의 토대가 되고, AI는 이 모든 과정을 가속화하고 있으며, 단백질 구조를 밝혀내는 것은 이 패러다임이 시작되는 출발점이 되는 것이다.






최근댓글